Research Projects
Genome sequences of bacteriophages that infect Salmonella Typhi from Bangladesh
Abstract
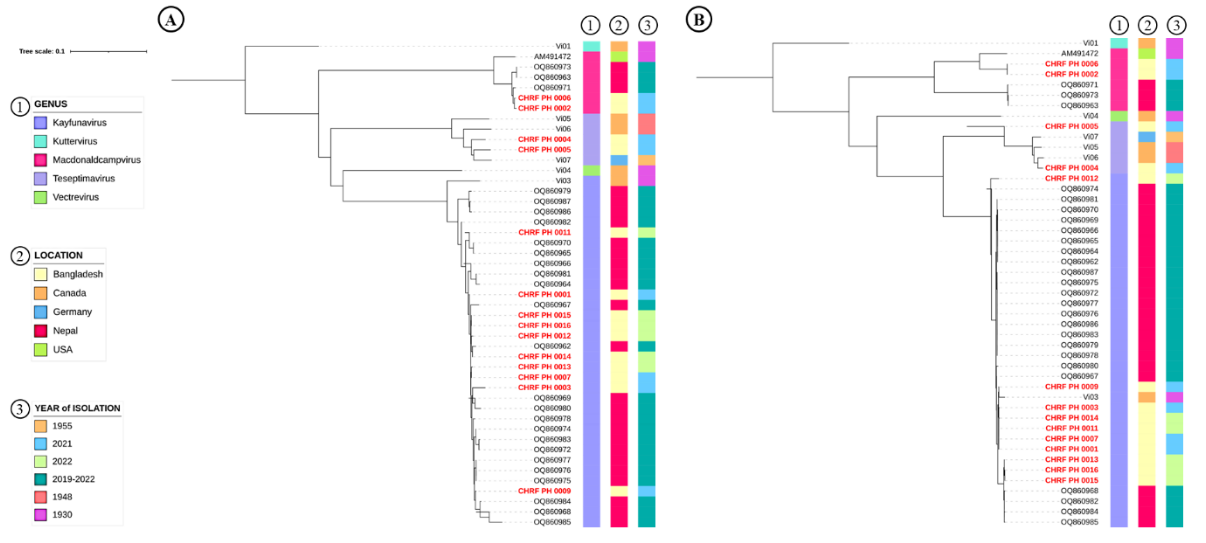
This report presents the near-complete genome sequences of 14 bacteriophages that infect Salmonella Typhi, identified through environmental surveillance in Bangladesh between August 2021 and June 2022. The bacteriophages, belonging to the genera Kayfunavirus, Macdonaldcampvirus, and Teseptimavirus, share evolutionarily ties with previously documented Typhi bacteriophages.
Keyword:
Funding: This study was funded by the Bill and Melinda Gates Foundation (INV-003717) .
Parv4 detection in children with suspected meningitis is associated with high mortality.
Abstract
Bacteriophages (or phages) are viruses that infect bacteria and regulate their abundance, phenotypic characteristics, and long-term evolutionary trajectory. Recently, our group has shown that Typhi-specific phages are abundant in surface water collected from high typhoid burden settings in Bangladesh and phage abundance correlates with typhoid burden. However, no information is available regarding the infecting mechanisms of Typhi-phages and the role these phages play in the evolution and dynamics of Salmonella Typhi in the environment. In this study, we used a combination of whole genome sequencing and bacterial killing assays against a diverse panel of Salmonella Typhi isolates to characterize the Typhi-phages present in Bangladesh.
Keyword: Water-borne diseases, factors, vibrio cholerae, public health, health impacts, calamities, water.
On review
Global Pneumococcal Sequencing Project.
Abstract
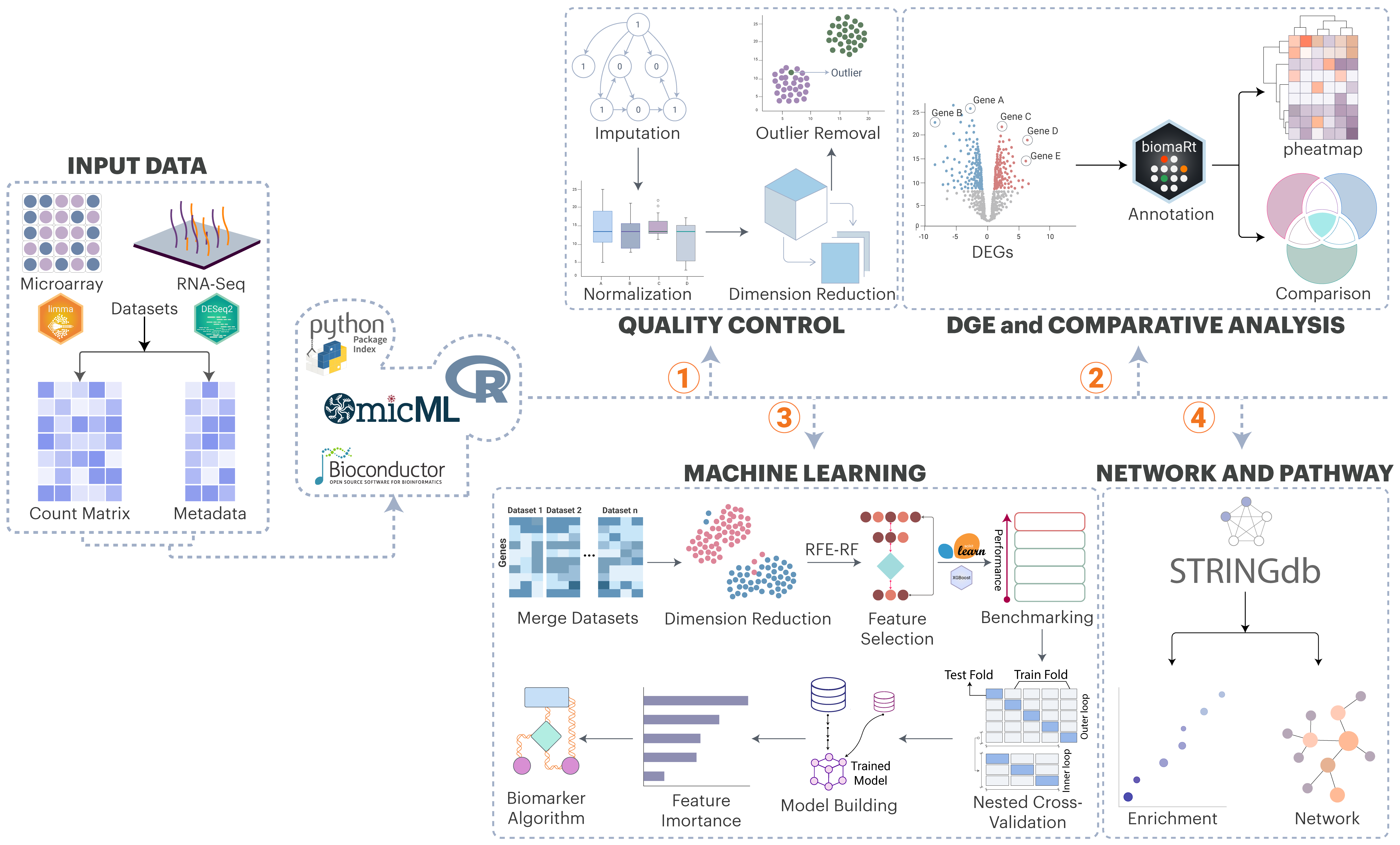
omicML: An Integrative Bioinformatics and Machine Learning Framework for Transcriptomic Biomarker Identification
Abstract
Transcriptomic biomarker discovery has been a challenge due to variation in datasets and platforms, complexity in statistical and computational methods, integration of multiple programming languages, and intricacy of ML workflow to evaluate biomarkers. Standard workflows necessitate several stages (quality control, normalization, differential expression), typically executed in R or Python, resulting in bottlenecks for non-experts. Existing platforms have alleviated certain challenges by offering graphical interfaces for data loading, normalization, differential gene expression analysis, and functional analysis; nevertheless, they typically do not incorporate integrated machine learning procedures for biomarker selection.
Keywords: GUI, DGE, DEGs, LFC, FDR, Padj, PCA, UMAP, t-SNE, ML, LR, ET, RF, XGB, GB, AB, ACC, BACC, PREC, REC, F1, AUROC, AUPRC, MCC, KAPPA, LOGLOSS, Mpox, MPXV, GEO
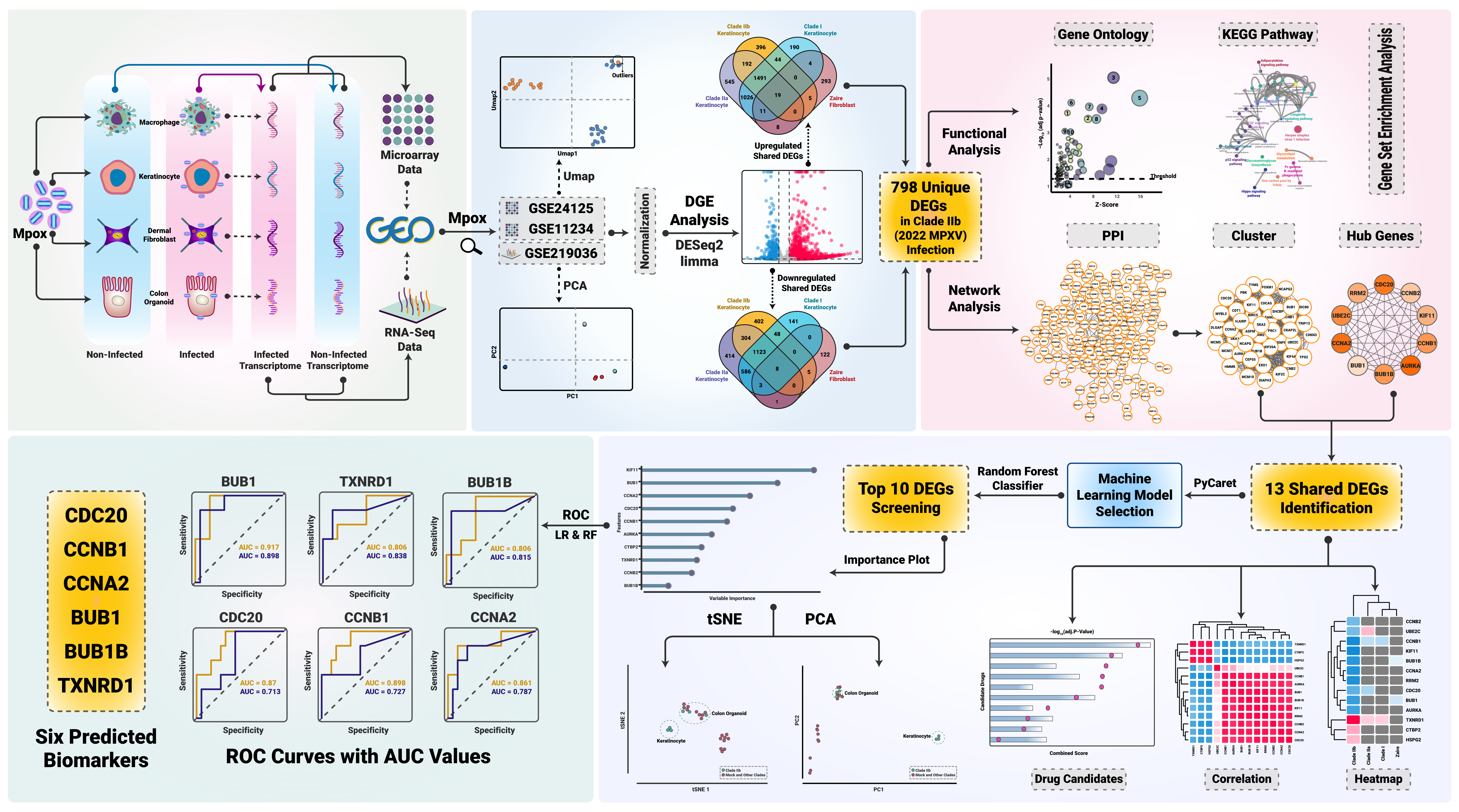
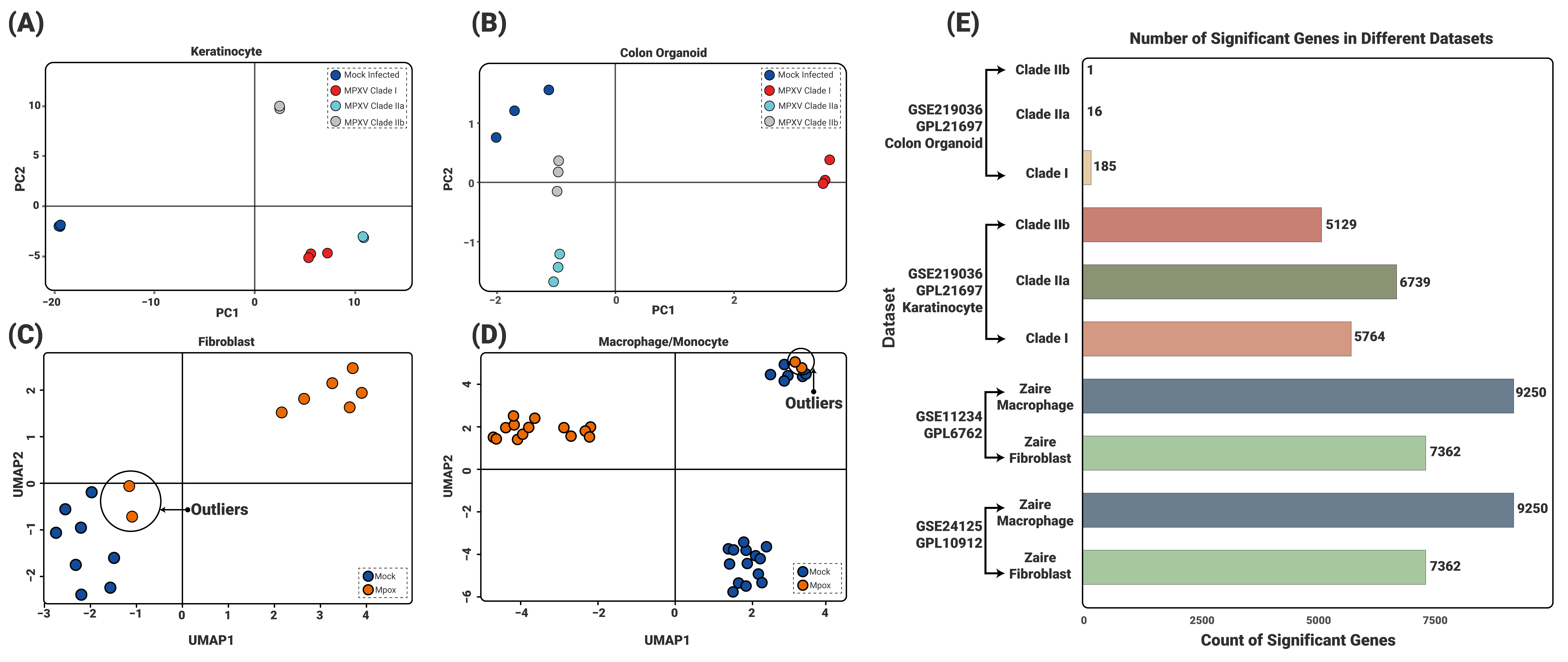
Identification of Potential Biomarkers for 2022 Mpox Virus Infection: A Transcriptomic Network Analysis and Machine Learning Approach
Abstract
Monkeypox virus (MPXV), a zoonotic pathogen, resurged in 2022 with the Clade IIb variant, raising global health concerns due to its unprecedented spread in non-endemic regions. Recent studies revealed that Clade IIb (2022 MPXV) is characterized by unique genomic mutations and epidemiological behaviors, suggesting variations in host-virus interactions. This study aimed to identify differentially expressed genes (DEGs) induced by the 2022 MPXV infection through comprehensive bioinformatics analyses of microarray and RNA-Seq datasets from post-infected cell lines across different MPXV clades. Gene expression network analyses pinpointed key DEGs, followed by candidate drug assessment using the Drug SIGnatures DataBase (DSigDB) and validation by multiple machine learning models. Comparative differential gene expression (DGE) analysis revealed 798 DEGs exclusive to the 2022 MPXV invasion in skin cell lines (keratinocytes and fibroblasts). Intriguingly, 13 key DEGs were identified across hubs and clusters, highlighting their aberrant expression in cell cycle regulation, immune responses, and cancer pathways. Biomarker screening via a Random Forest (RF) model (selected with PyCaret from multiple models) and validation through t-distributed stochastic neighbor embedding (t-SNE) algorithm, principal component analysis (PCA), and ROC curve analysis employing Logistic Regression and Random Forest identified 6 key DEGs (TXNRD1, CCNB1, BUB1, CDC20, BUB1B, and CCNA2) as promising biomarkers (AUC > 0.7) for Clade IIb infection. This study anticipates that further investigation and clinical trials will catalyze the development of novel detection and therapeutic options to combat the 2022 MPXV infection in humans.
Keywords: Mpox (monkeypox), 2022 MPXV (Clade IIb), DEGs, machine learning (ML) models, biomarker, candidate drugs
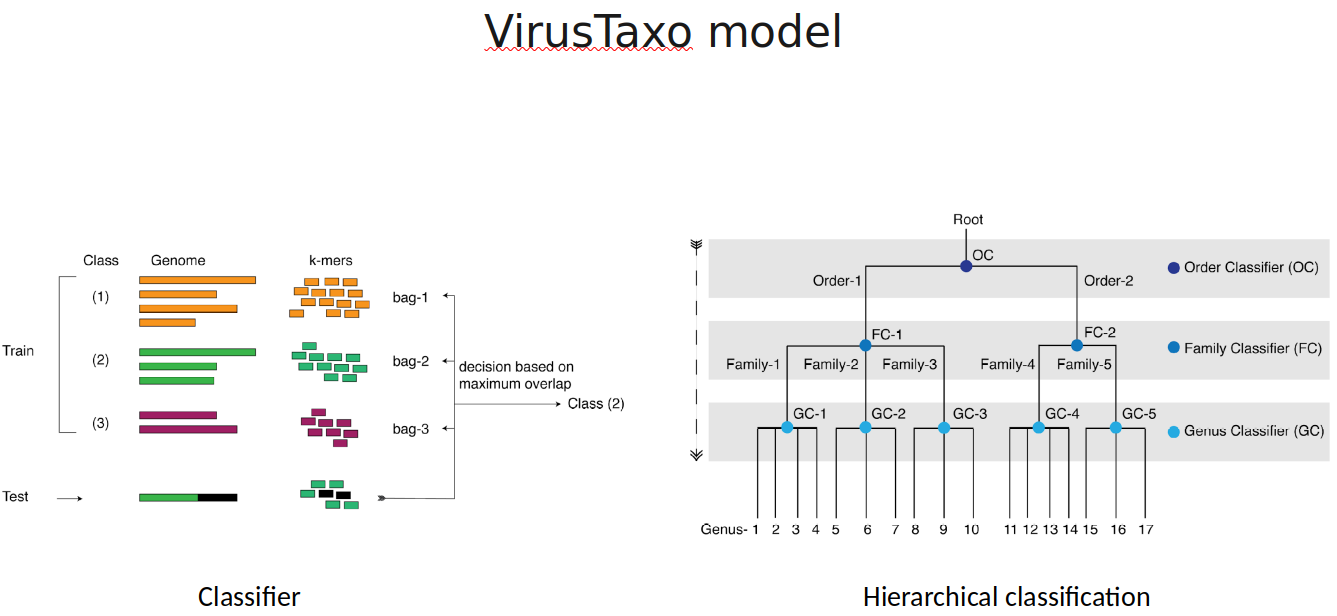
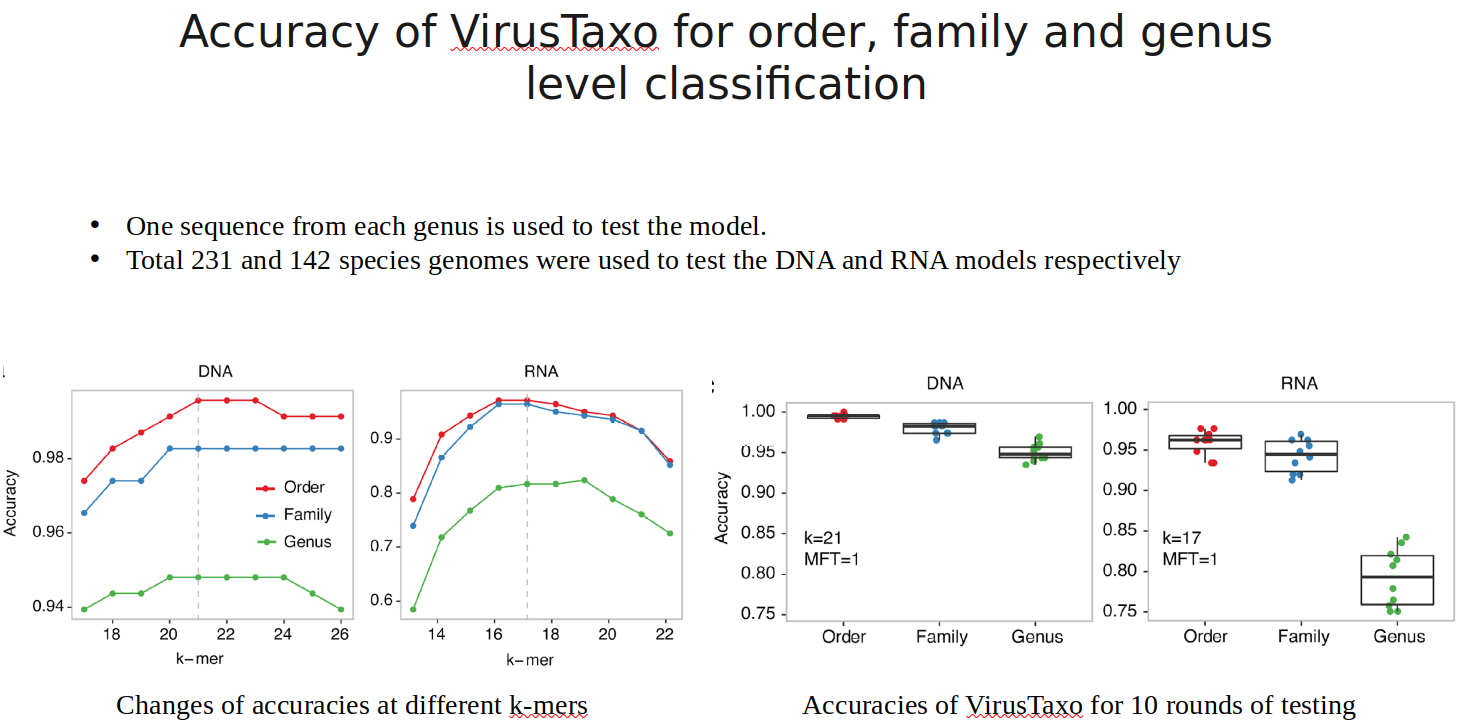
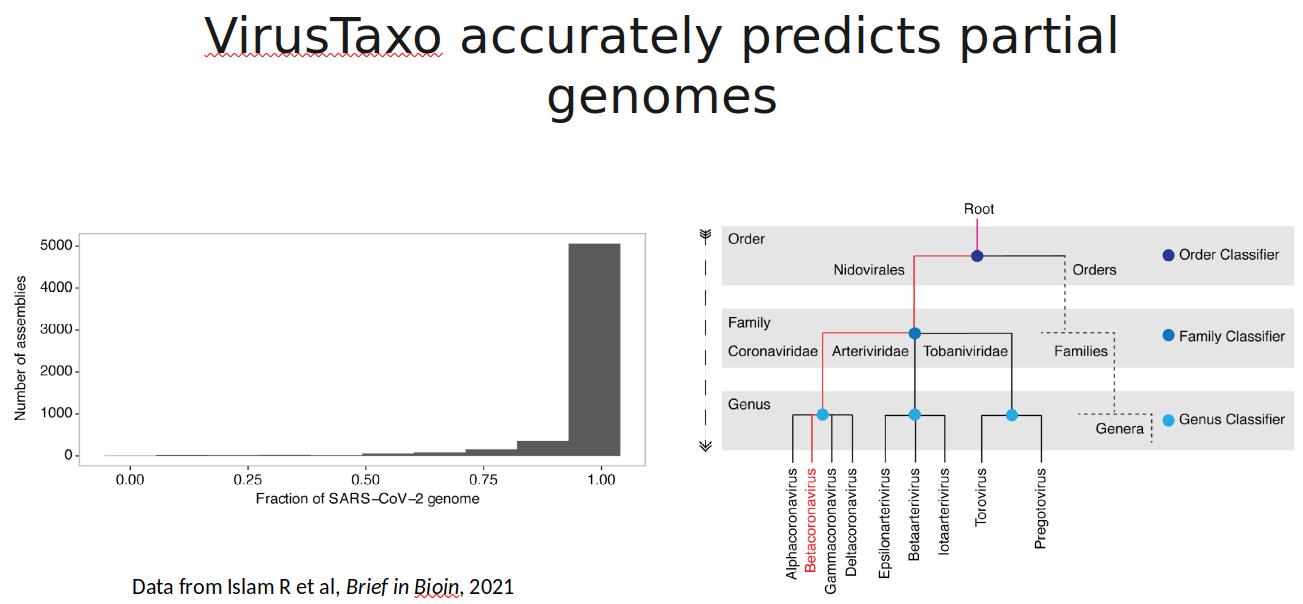
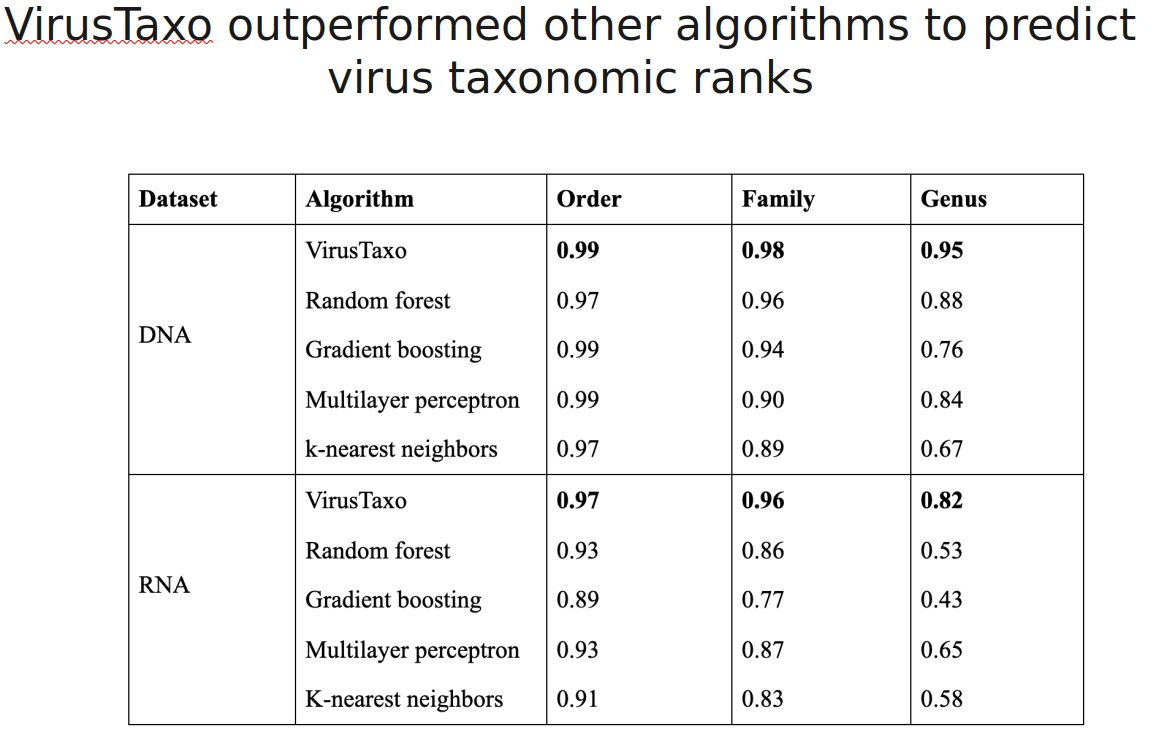
VirusTaxo: Taxonomic classification of viruses from the genome sequence using k-mer enrichment
Abstract
Classification of viruses into their taxonomic ranks (e.g., order, family, and genus) provides a framework to organize the vast population of viruses. Next-generation metagenomic sequencing technologies have led to a rapid increase in viral sequencing data, necessitating the development of bioinformatics tools to analyze viral taxonomy. While many metagenomic taxonomy classifiers have been created to study microbiomes, classifying the diverse range of virus sequences remains a significant challenge. There is a growing demand for specialized methods optimized for the classification of viral sequences into their respective taxa. To address this, we developed VirusTaxo, a tool for the taxonomic classification of viruses from metagenomic sequences, utilizing a diverse set of viral genera (e.g., 402 DNA and 280 RNA genera). VirusTaxo achieves an average accuracy of 93% at the genus level in predicting DNA and RNA viruses. The tool outperforms existing virus taxonomic classifiers by assigning taxonomy to a larger fraction of metagenomic contigs compared to other methods. Benchmarking VirusTaxo on a collection of SARS-CoV-2 sequencing libraries and metavirome datasets demonstrates that it can accurately characterize viral taxonomy from highly diverse contigs, providing reliable decisions on viral taxonomy.
Keywords: Virus, Taxonomy, Hierarchical classification, k-mer, Genome

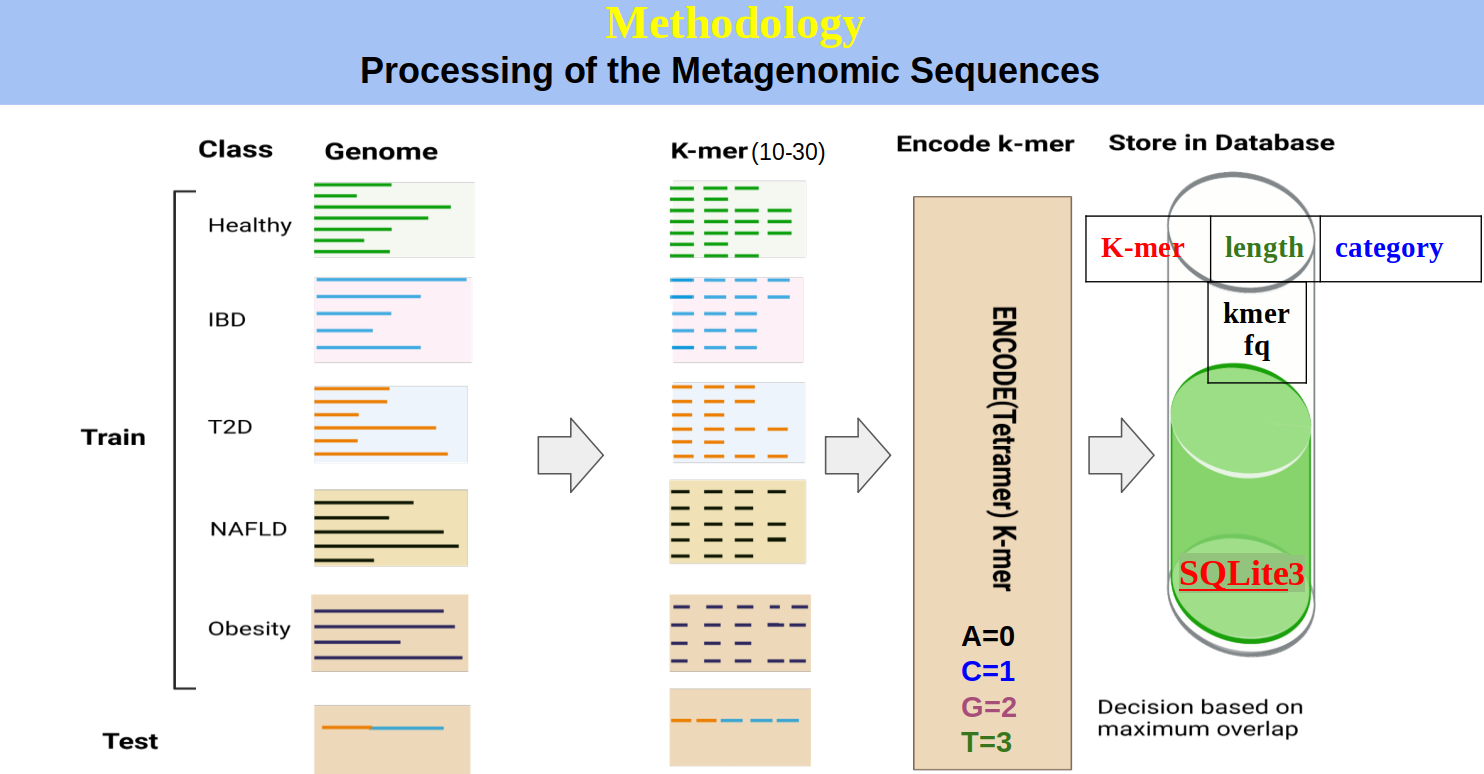
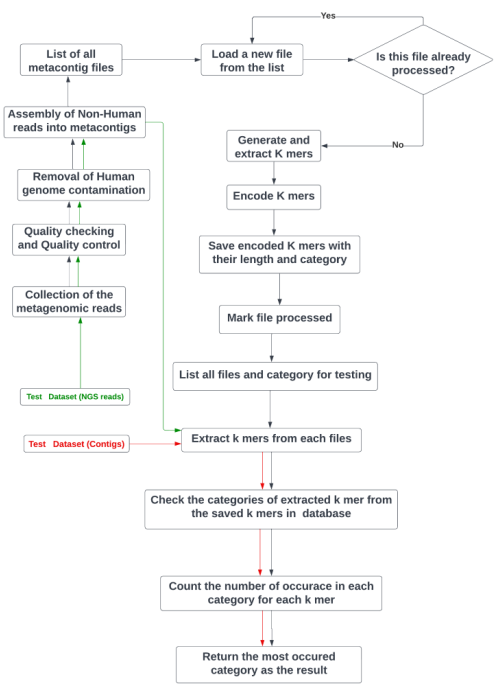
Development of a disease detection tool from shotgun metagenomic data utilizing algorithms based on K-mer frequency: A gut microbiome study.
Abstract
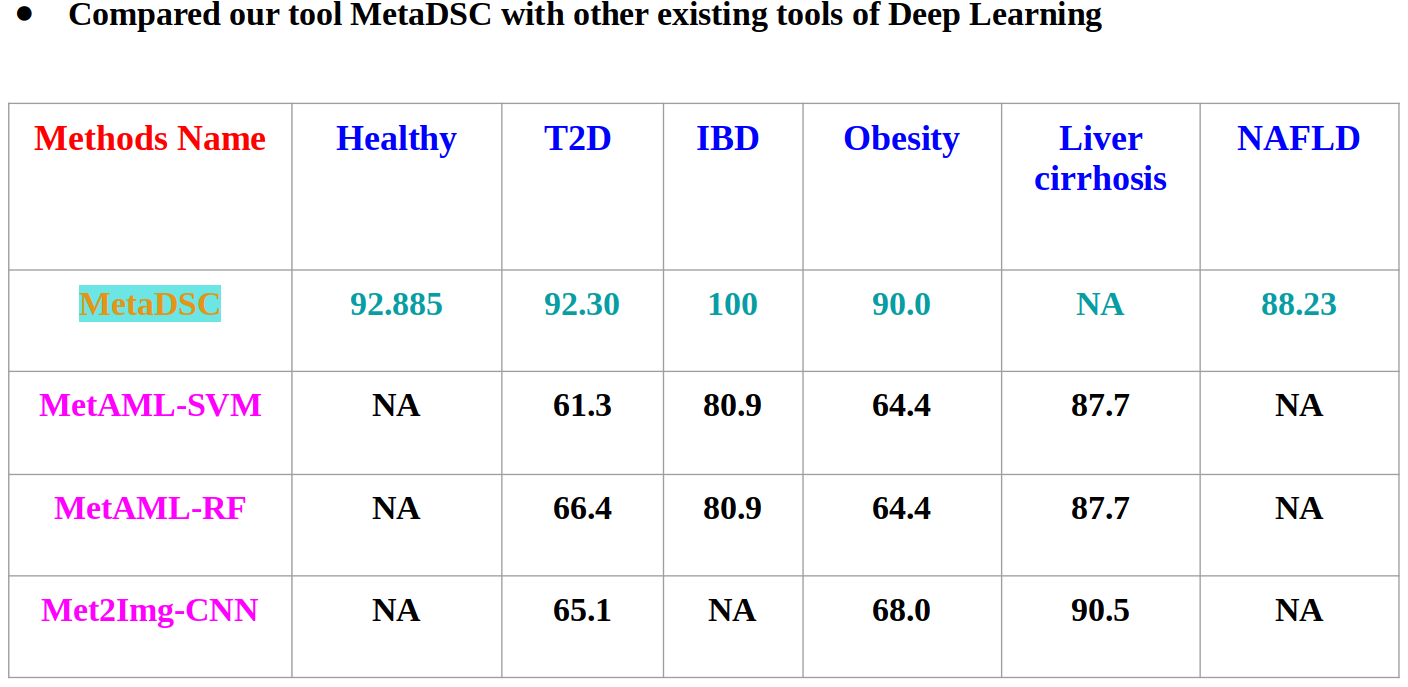
Human gut microbiome composition can be influenced by various factors, including dietary habits, lifestyle, ethnicity, geographic location, and many others. The gut microbiome plays a critical role in the development of various Non-Communicable Diseases (NCDs). Prediction of NCDs can be achieved by analyzing the gut microbiota. Using data from metagenomic sequencing in a multi-layered approach, it is possible to forecast a patient’s current state of health. Deep learning, a powerful form of machine learning, has been successfully applied in numerous biological fields by researchers. In this study, we developed a disease detection tool called the Metagenomic Disease-specific Classifier (MetaDSC), which employs algorithms based on the frequency of k-mers. MetaDSC was trained using whole genome shotgun sequencing data from different health conditions. When it comes to distinguishing between sick and healthy samples, MetaDSC achieves an accuracy rate that averages up to 92 percent. MetaDSC outperformed several existing tools in this regard. Specifically, MetaDSC can recognize Healthy, Type 2 Diabetes Mellitus (T2D), Non-Alcoholic Fatty Liver Disease (NAFLD), Inflammatory Bowel Disease (IBD), and Obesity from metagenomics sequences with 92.8%, 92.8%, 88.22%, 100%, and 90.0% accuracy respectively. MetaDSC has the potential to be utilized for accurate diagnosis across a wide variety of diseases, provided that new datasets are consistently incorporated into the tool.
Keywords: Gut Microbiome, Non-Communicable Diseases, Metagenomic, Machine learning , Deep learning, k-mer, MetaDSC.

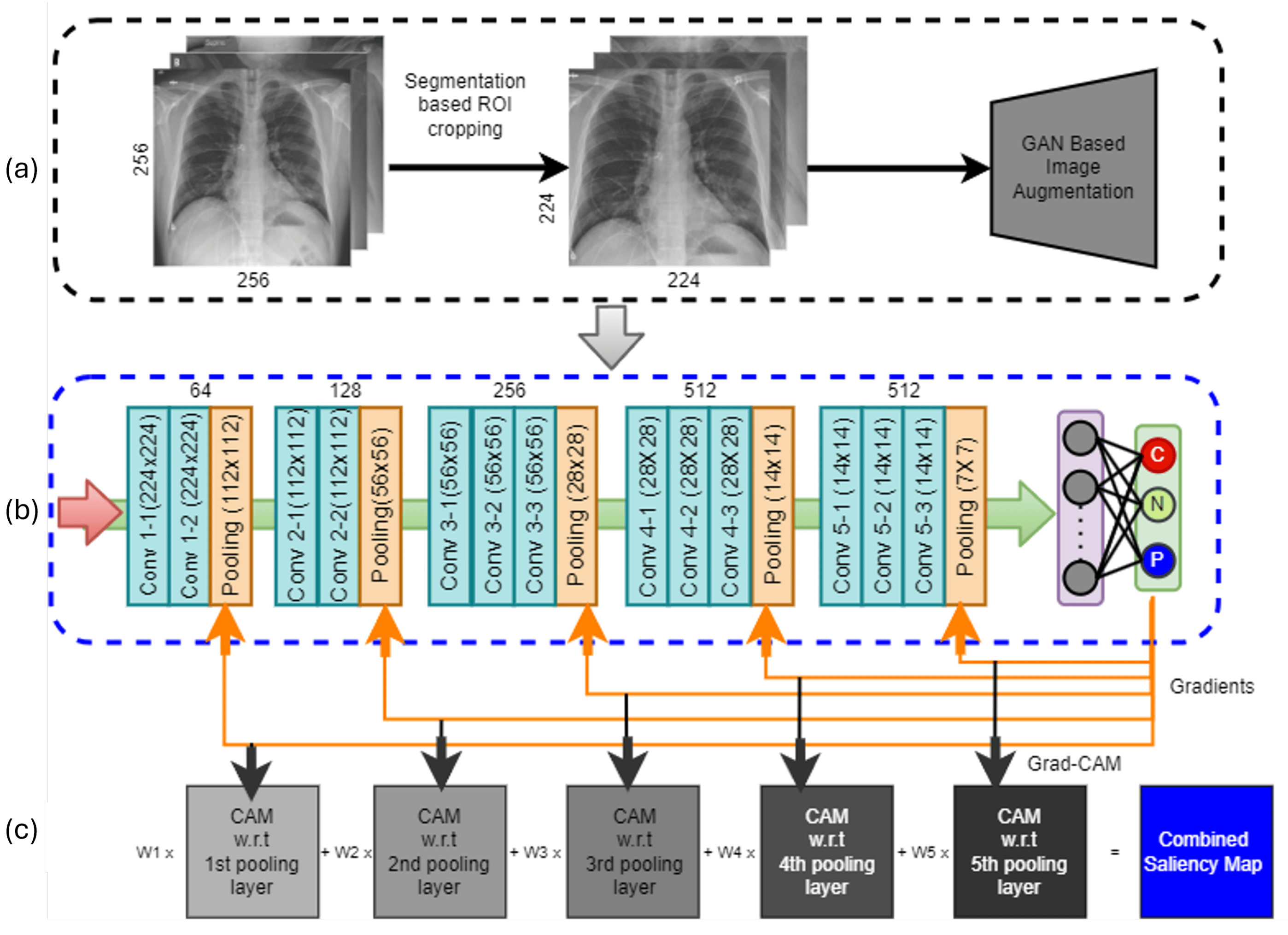
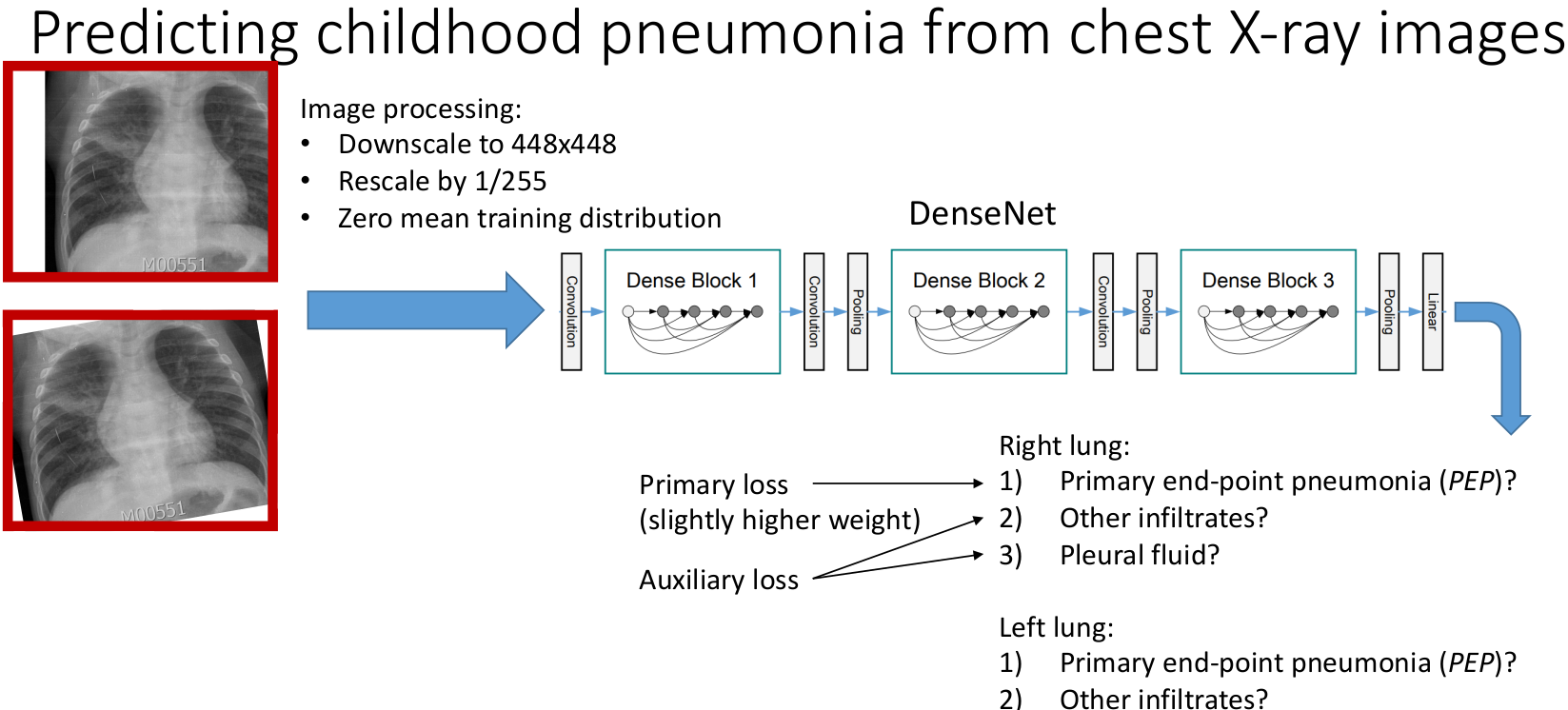
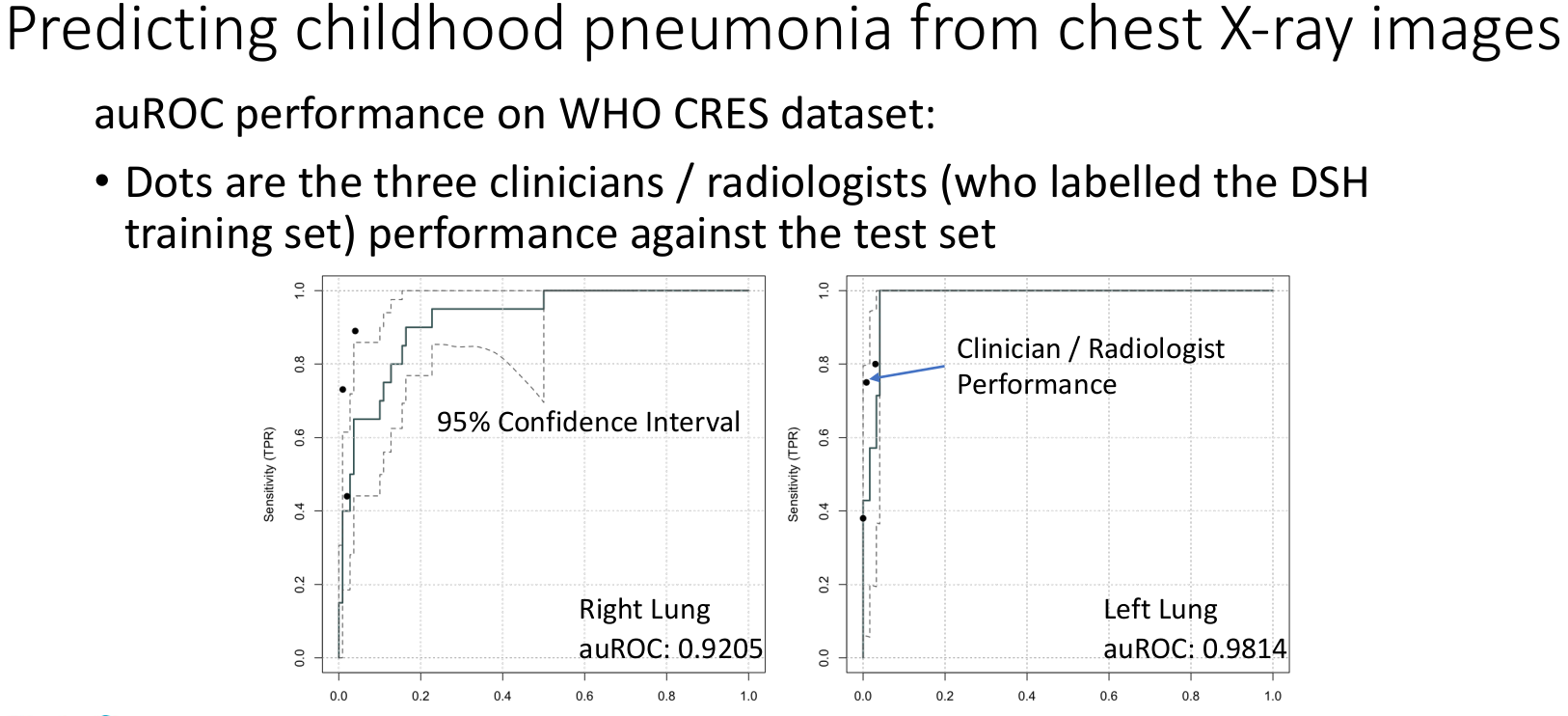
Computational framework to interpret chest X-rays and diagnose pneumonia
Abstract
In low- and middle-income countries, pneumonia remains the leading cause of illness and death in children under 5 years. The recommended diagnostic tool for pediatric pneumonia is chest X-ray image interpretation, which is challenging to standardize and requires trained clinicians or radiologists. Current automated computational tools predominantly focus on assessing adult pneumonia and have been trained on images evaluated by a single specialist. This study aims to develop a computational tool using a deep learning approach to diagnose pediatric pneumonia from X-ray images assessed by multiple specialists trained by the WHO expert X-ray image reading panel.
Workingg to the previous extended project
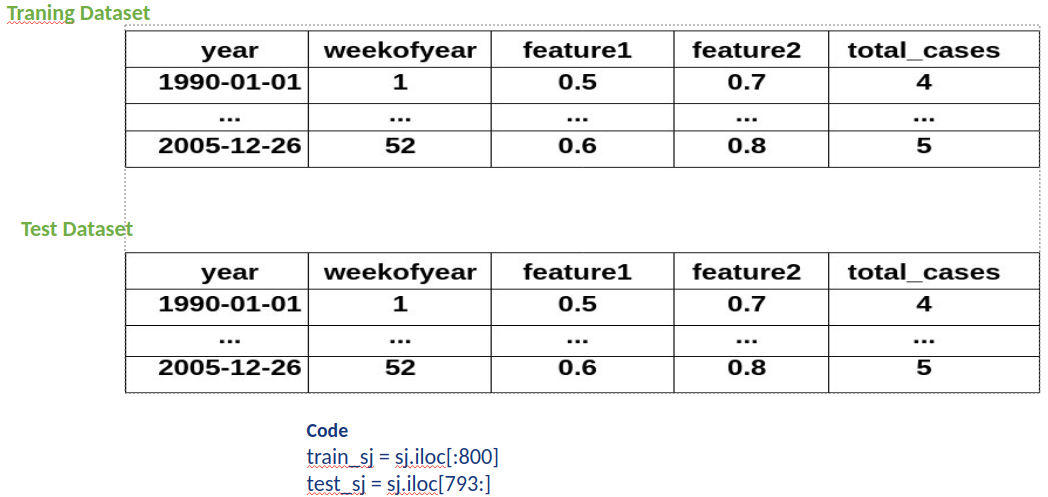
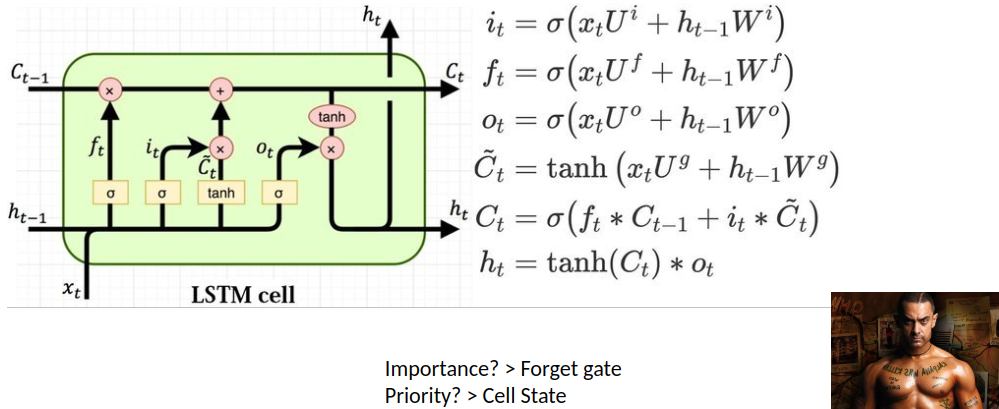
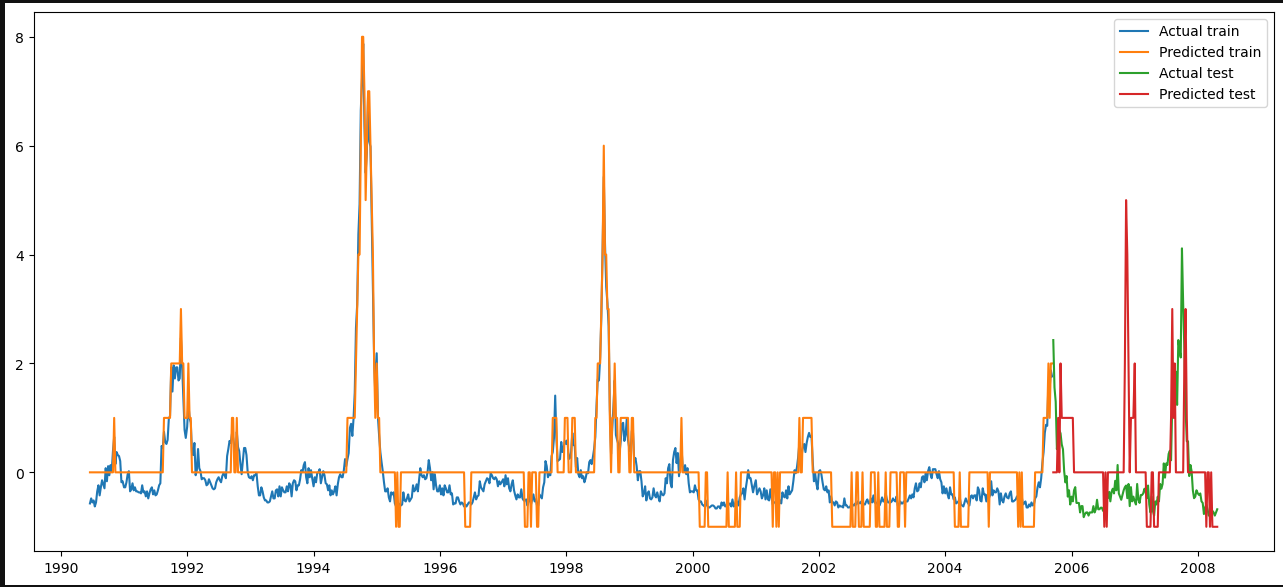
Predicting Disease Spread of Dengue using LSTM
Abstract
Mosquito-borne dengue fever is a disease found in tropical and subtropical regions of the world. In mild cases, fever, rash, and sore muscles and joints resemble flu symptoms. serious dengue fever can result in hypotension, serious bleeding, and even death.
Dengue transmission dynamics are influenced by climate variables like temperature and precipitation since the virus is spread by mosquitoes. Despite the complexity of the relationship, an increasing number of scientists contend that changes in distribution brought about by climate change would likely have a major impact on public health globally.
Dengue fever has been increasing in recent years. Southeast Asia and the Pacific islands have historically had the highest rates of the illness.

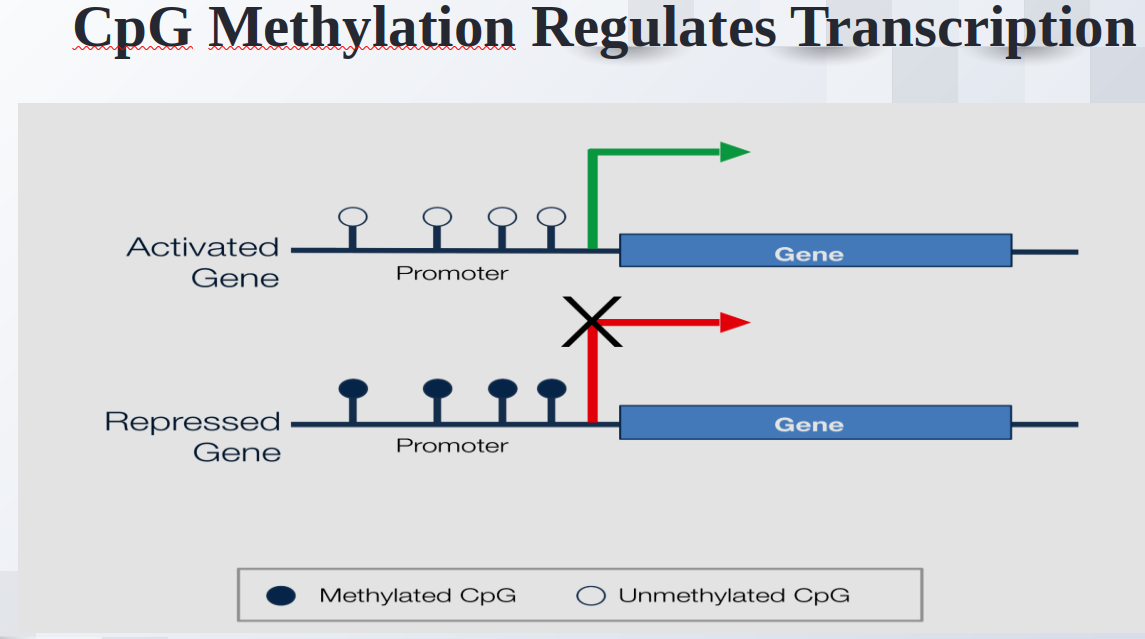
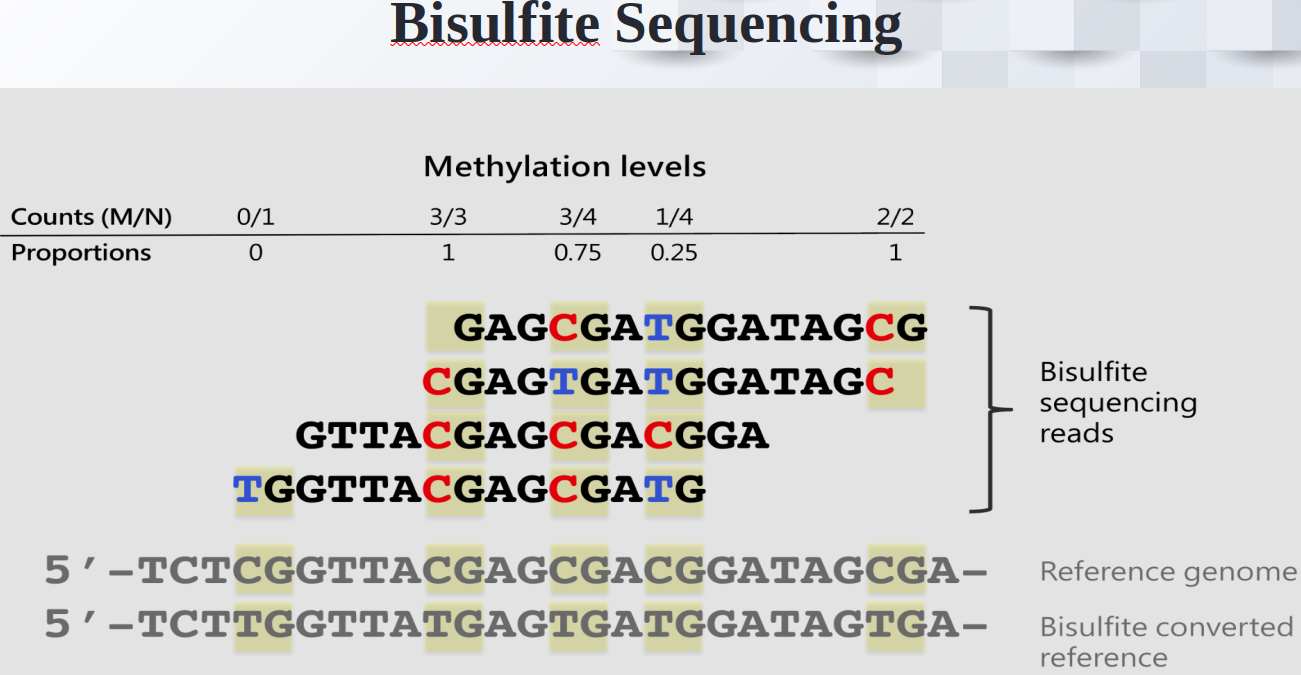
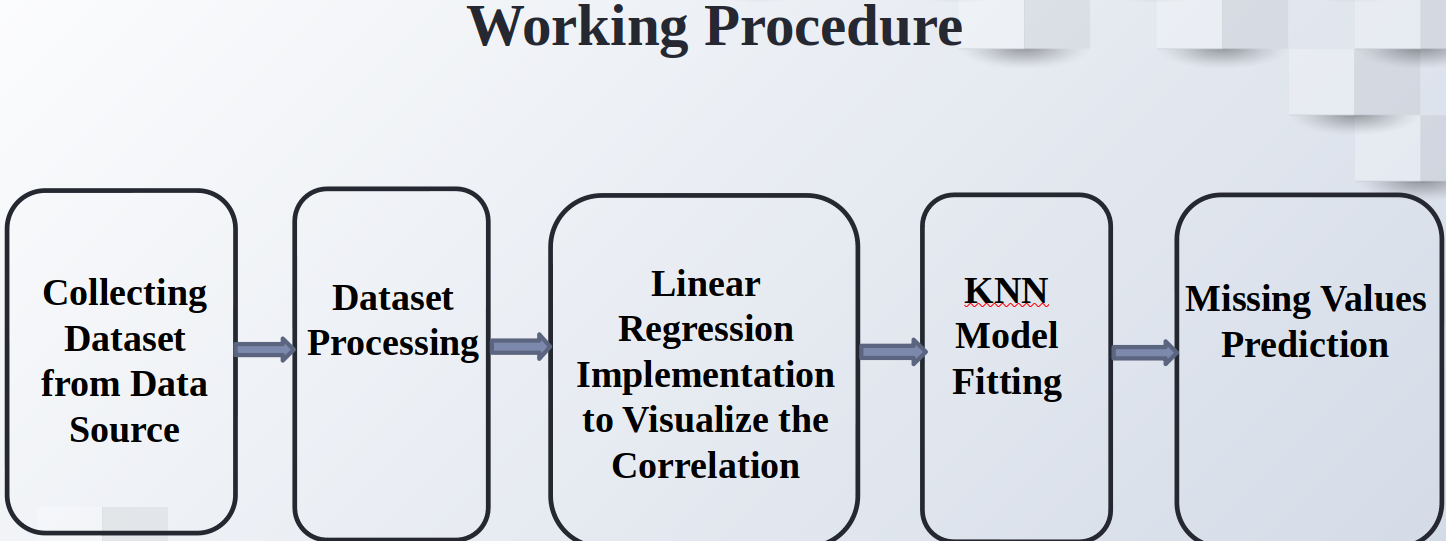
Prediction of Missing DNA Methylation fromWhole Genome Bisulfite Data Using KNN
Abstract
One important epigenetic alteration that is essential for controlling gene expression and other biological functions is DNA methylation. One effective method for single-base resolution DNA methylation profiling is Whole Genome Bisulfite Sequencing (WGBS). However, missing methylation values in WGBS datasets are frequently the consequence of technical difficulties and experimental limitations. It is imperative to address these absent values in order to gain a thorough understanding of the epigenetic landscape. In this work, we suggest a novel method for predicting missing DNA methylation values in WGBS data, which is based on K-Nearest Neighbours (KNN). Our approach accurately imputes missing values by utilising the methylation data’s inherent structure and patterns. KNN, an instance-based, non-parametric machine learning algorithm, is used to find comparable methylation profiles for each
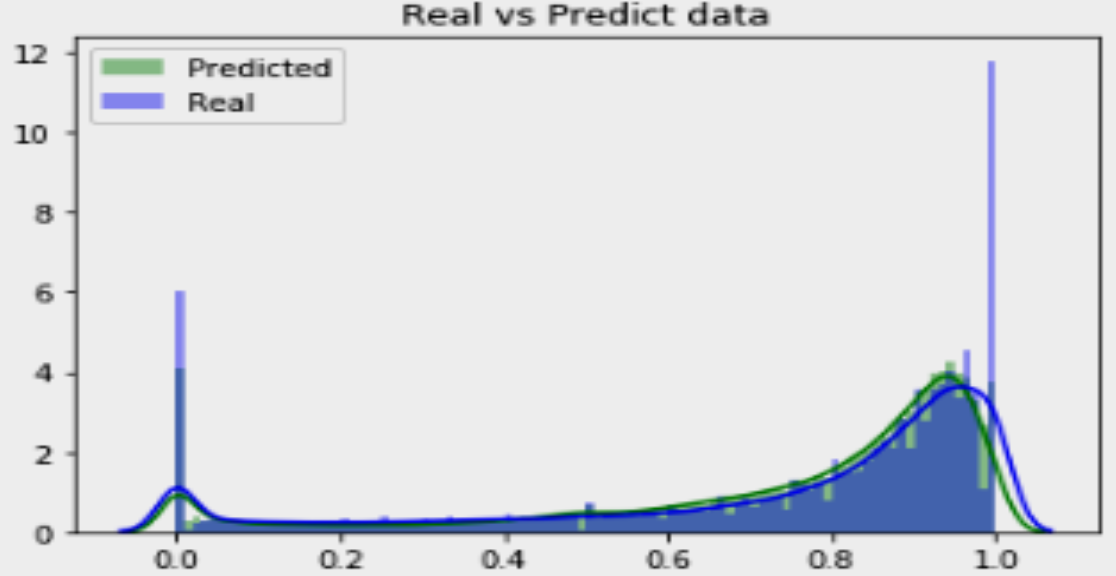
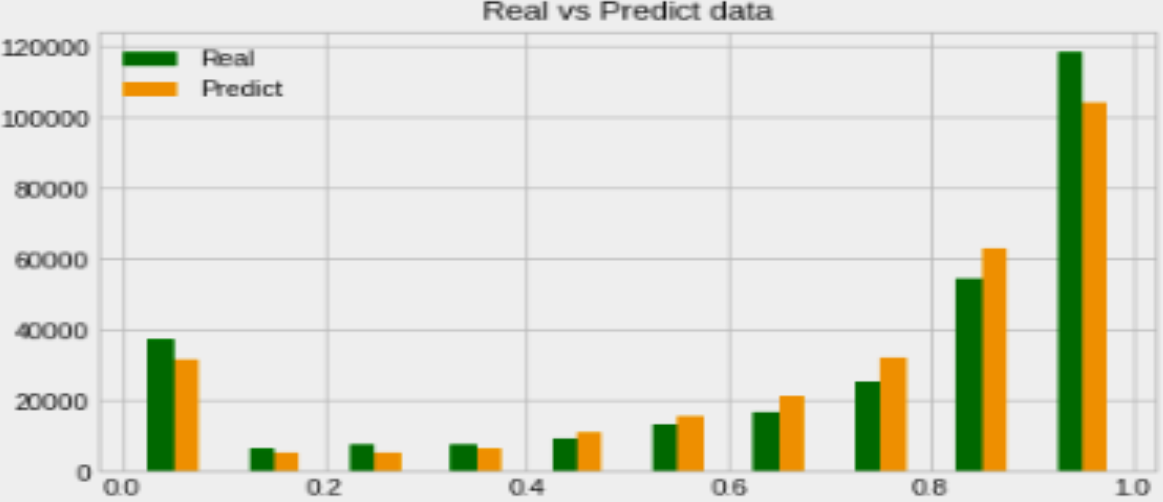
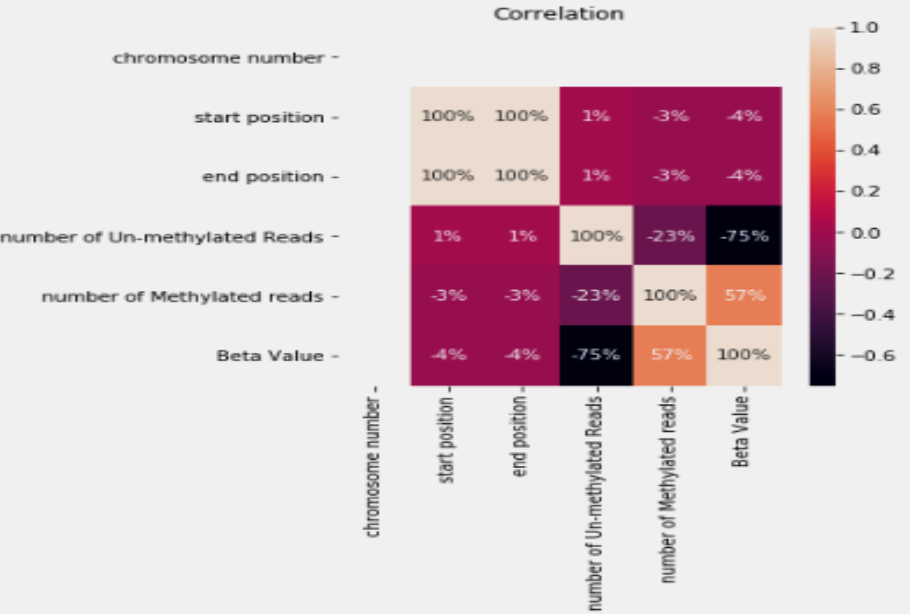
Title of the Video Goes Here
A Global Pediatric Cell Atlas of Nasal and Oral Mucosa
Abstract
The nasopharyngeal and oral mucosa represent the initial sites of interaction with many environmental agents and microbes. Recent single-cell studies have revealed a rich diversity of epithelial and immune cell types and states within nasopharyngeal epithelium in diseases of global significance, including allergic inflammation and viral infection. Yet, beyond an accessible window into disease biology, minimally-invasive sampling of the nose and mouth in children represents a truly unique opportunity to characterize healthy mucosal epithelial and immune function worldwide. However, a comprehensive map of epithelial and immune system development across diverse ancestries and environments is lacking. To more broadly investigate the nasal and oral mucosa and understand how the normal variation present in healthy children maintains health or may inform disease, we have assembled an interdisciplinary team: unifying experts in 7 cities in 5 countries who are deeply invested in understanding the single-cell biology of the nasopharyngeal and oral mucosa in children living within our communities. Our plan aims to generate scientific and community engagement in all phases of our research to establish the foundation in Years 1 and 2 that will enable us to carefully and considerately analyze 80 pediatric participants at each site (560 total) across the age range from 1-month to 18-years of age in the next phase of our network (potential Year 3 and beyond). Our team will pilot and analyze single-cell data jointly with scientists from all locations, and share important lessons in global science, protocols and resultant data openly with the community. Ultimately, our global single-cell based characterization of the developing nasopharyngeal mucosa will reveal principles of epithelial and immune system development that will facilitate the equitable development of novel therapies for diseases of the aerodigestive tracts.
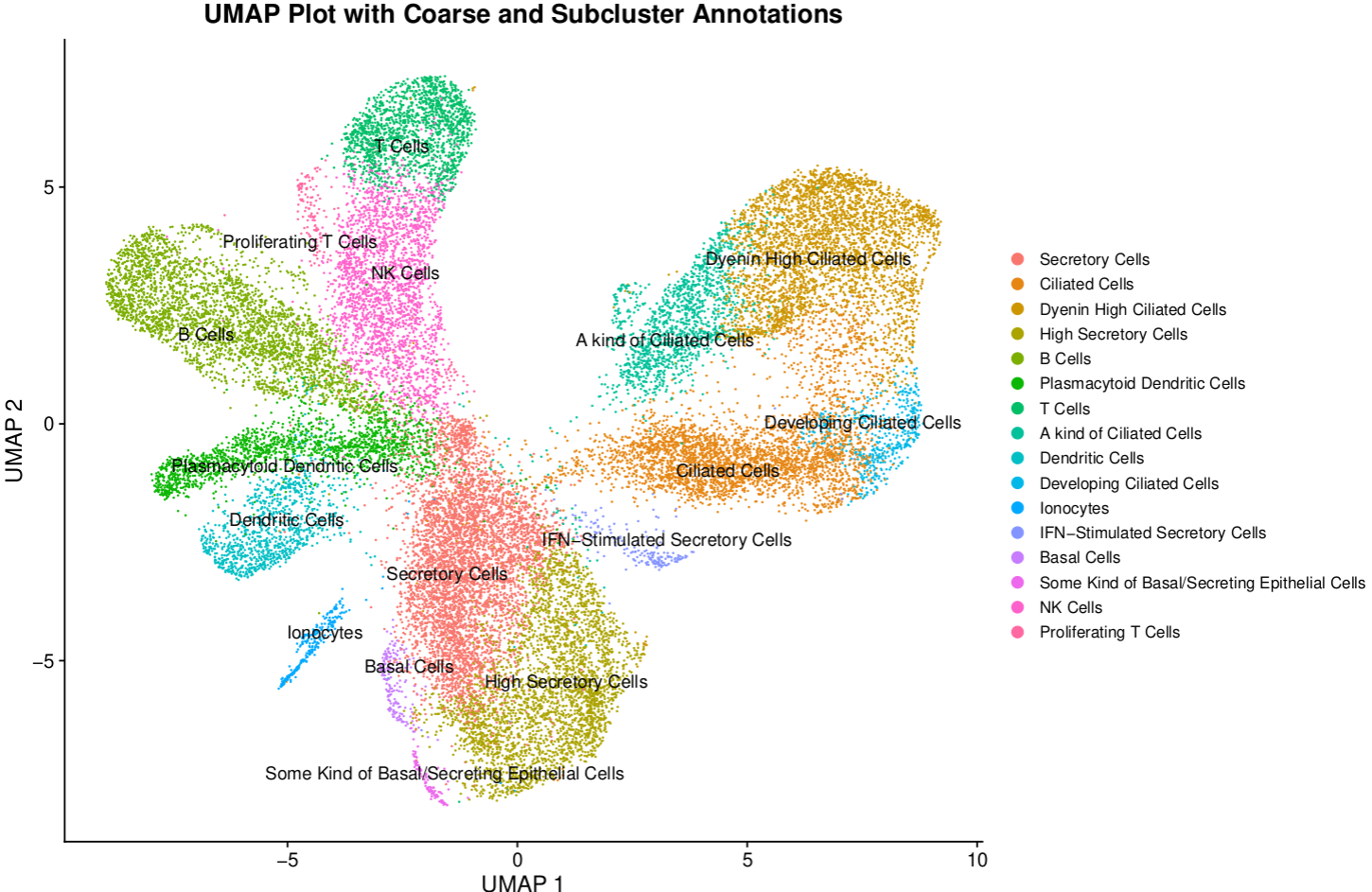
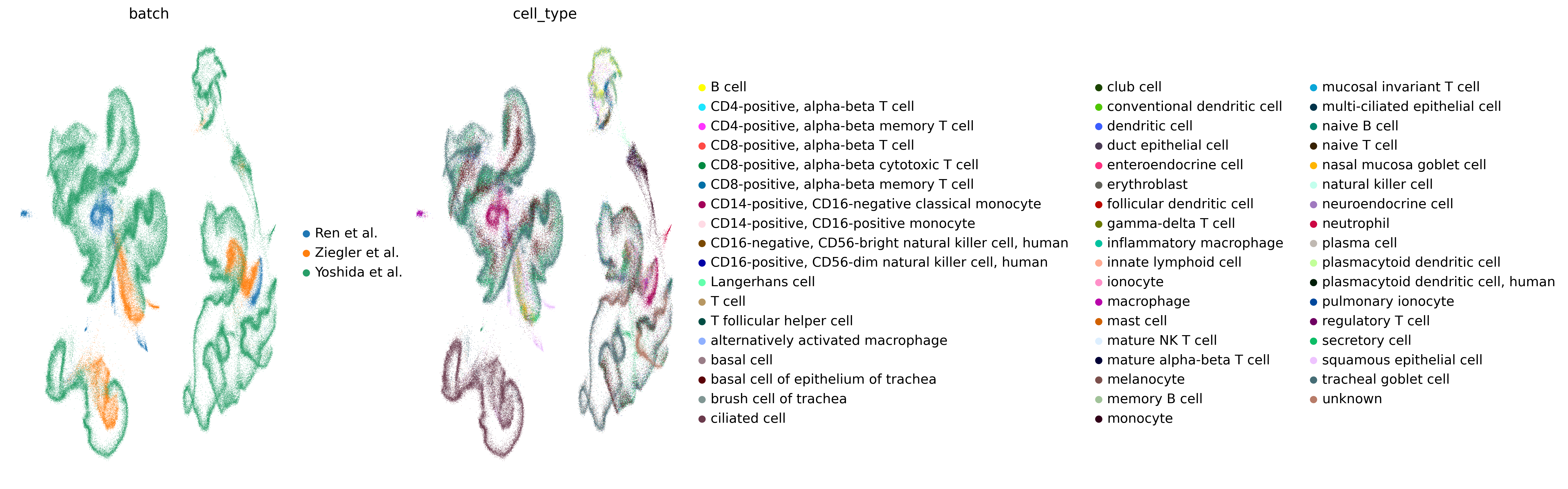
Building a Single‑Cell Atlas of the Nasopharyngeal Mucosa to Investigate SARS‑CoV‑2 Infection
Abstract
Steps in Building the Atlas:
- Dataset Selection: Tissues related to the respiratory system, focusing on the nasopharynx.
- Integration of Datasets: From studies by Yoshida, Ziegler, and Ren, using Harmony to combine over 245,000 cells.
- Clustering Analysis: Of the integrated dataset to define distinct cell types in the nasopharyngeal tissue.
- Identification of Cell-Type Clusters: With the integrated dataset and visualization of immune cell subtypes.
- SARS‑CoV‑2 Expression Assessment: In the nasopharynx.
- Reannotation of Cell Types: To identify novel or other subtypes.
- Demographic Analysis: Analyze the dataset with demographic details like age, SARS-CoV-2 status, and more.
- Comparative Analysis: Compare findings from the Bangladesh cohort with the global integrated dataset.
PCV effectiveness study via single‑cell analysis of pregnant women.
Abstract
A single-cell analytics platform to track the immune responses of babies before and after receiving a pneumococcal conjugate vaccine to determine the impact of various factors, including nutritional status and seasonality, on vaccine efficacy. Vaccines have successfully reduced childhood morbidity and mortality; however, their efficacy can be influenced by host factors and extrinsic factors through unknown cellular mechanisms. They will recruit 50 newborns in a rural district north of Dhaka and collect blood and nasopharyngeal swabs before, during and after a routine vaccination series. They will extract peripheral blood mononuclear cells and use them to perform single-cell RNA sequencing to identify cell subtypes and link differential vaccine responses to factors including gestational age, nutritional status and sex.
RSV Vaccine Impact Monte Carlo Simulation
Abstract
Updating